We develop and apply a variety of computational methods, including: atomistic simulation, density functional theory and structure prediction to investigate the materials’ structure-property relation, as well as Datahub for new materials discovery.
Our goal is to discover and design new materials in the following aspects:

Organic Crystal Polymorphism
Predicting the crystal structures for a given molecule could help determine the existence of different forms and suggest as yet unseen polymorphs of currently known structures.
Defects in Materials
Understanding the structural and functional properties influenced by defects is key to optimizing next-gen materials needed for advanced energy applications.
Topological phonon database!
The first online topological phonon database with over 5000 materials is in live!
Latest paper (2022/10/28)
Simulation of GaN’s phase transition using the machine learning metadynamics
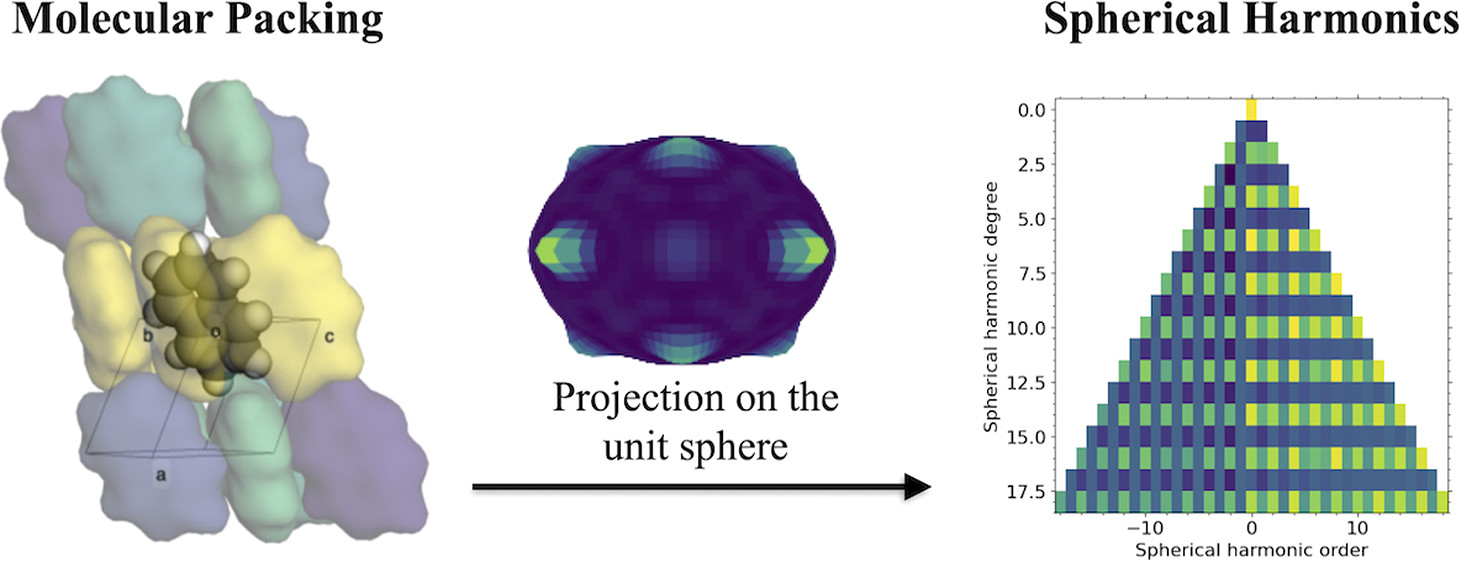
Latest paper (2022/10/27)
Quantification of Crystal Packing Similarity from Spherical Harmonic Transform