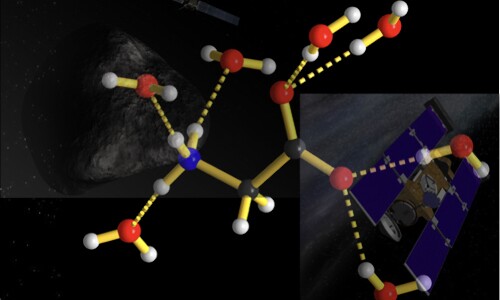
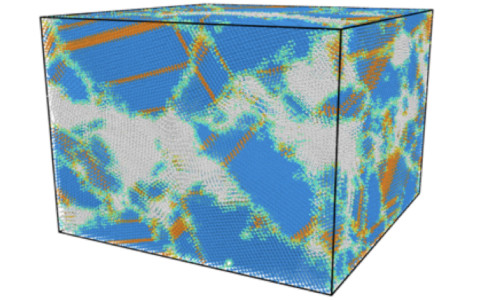
We develop and apply a variety of computational methods, including: atomistic simulation, density functional theory and structure prediction to investigate the materials’ structure-property relation.
Our goal is to discover and design new materials in the following aspects: