Introduction
Trial-and-error experiments have dominated the discovery of new materials in the past. In the information age, tremendous efforts have been devoted to shifting this paradigm to rational strategies guided by virtual screening, which requires the development of new advanced modeling methods. To speed the discovery of new materials from computer modeling, our group has been focusing on two aspects of materials informatics (1) developing the accurate machine learning potentials and (2) construct the functional materials databases.
Machine Learining Interatomic potentials
Atomistic simulations have been used routinely to model the physical behaviors of many complex systems. The accuracy of the simulations is highly dependent on the underlying potential energy surface (PES) of the system. In principle, MD simulations can be based on quantum-mechanical or classical force field methods. The ab initio MD (AIMD) simulations usually employ density functional theory (DFT) approximation, which can provide a reliable representation of the system but is be limited to only a few hundreds of atoms. On the other hand, the classical MD method can model large systems at long-time scale, but they are often inadequate to describe the quantitative properties of the system.
Recently, machine learning methods have been widely applied to resolve the dilemma in comprising between accuracy and cost. The machine learning interatomic potential (MLIAP) are trained by minimizing the cost function to attune the model to deliberately describe the ab initio data. The cost of atomistic simulation is orders of magnitude lower than the quantum mechanical simulation, allowing the system to be scaled up to about one million atoms.
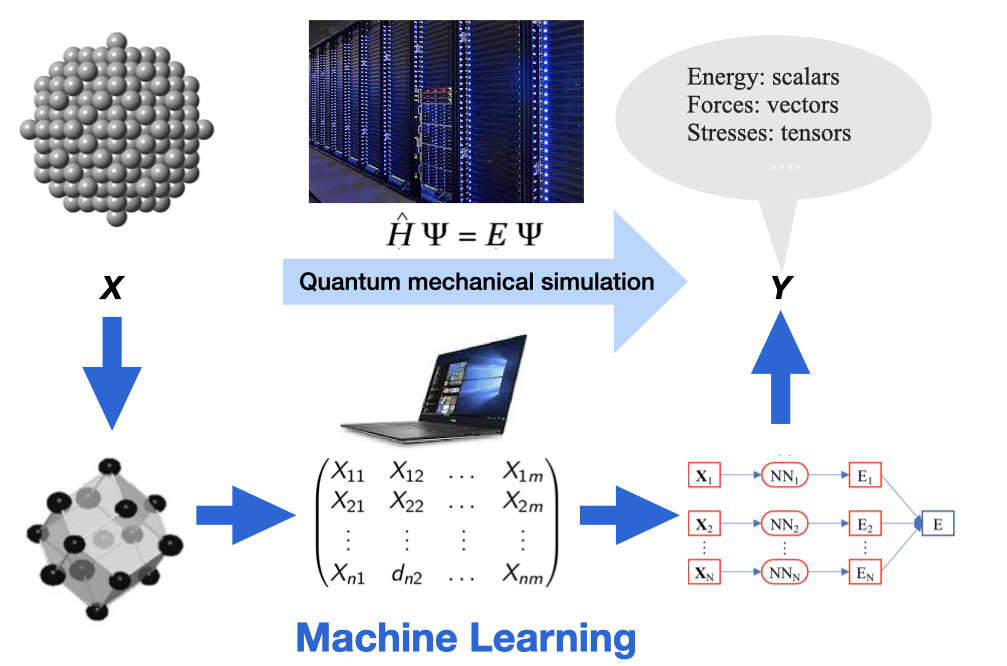
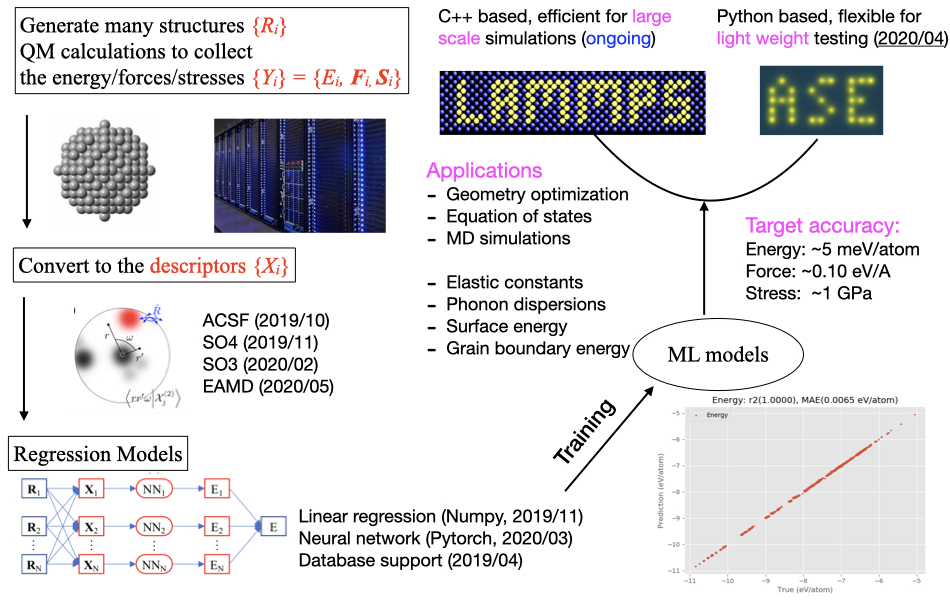
Since 2019, our group has been developing the PyXtal_FF —a package for generating machine learning potentials. The aim of PyXtal_FF is to promote the application of atomistic simulations by providing several choices of structural descriptors and machine learning regressions in one platform. Based on the given choice of structural descriptors (including the atom-centered symmetry functions, embedded atom density, SO4 bispectrum, and smooth SO3 power spectrum), PyXtal_FF can train the MLPs with either the generalized linear regression or neural networks model, by simultaneously minimizing the errors of energy/forces/stress tensors in comparison with the data from the ab-initio simulation. The trained MLP model from PyXtal_FF is interfaced with the Atomic Simulation Environment (ASE) package, which allows different types of light-weight simulations such as geometry optimization, molecular dynamics simulation, and physical properties prediction. Finally, we will illustrate the performance of PyXtal_FF by applying it to investigate several material systems, including bulk, surfaces and nanoparticles.

Relevant works
-
Yanxon H., Zagaceta D., Wood B.C., Zhu Q. (2020). Neural Networks Potential from the Bispectrum Component: A Case Study on Crystalline Silicon. (Open Access: PDF)
-
Zagaceta D., Yanxon H., Zhu Q. (2020). Spectral Neural Network Potentials for Binary Alloys. J. Appl. Phys., 128, 045113. (Open Access: PDF)
-
Yanxon H., Zagaceta D., Tang B., Matteson D., Zhu Q. (2020). PyXtal FF: a Python Library for Automated Force Field Generation. (Open Access: PDF)
-
Santos-Florez P. A., Yanxon H,Kang B, Yao Y-S, Zhu Q (2022). Size-Dependent Nucleation in Crystal Phase Transition from Machine Learning Metadynamics. Phys. Rev. Lett., 129, 185701 (Cover image) (arXiv: PDF)
Computational Materials Database
In the past, the identification of new materials required labor-intensive efforts, which included raw materials synthesis and sample characterization. Considering the current size of materials data (~30,000 after the removal of duplicates and structures with partial occupation) in the Inorganic Crystal Structure Database (ICSD), it is impossible to investigate each compound by trial-and-error experiments. The high-throughput (HT) computational materials design, based on a search for target materials from a large database containing necessary thermodynamic and electronic properties for all available materials, has become popular in materials science in recent years. Our group is generally interested in combinning the advanced structure prediction techniques with the high-throughput screening to discover the materials with target functionalities. Specifically, our efforts are focusing on the electrides and topological phononic materials.
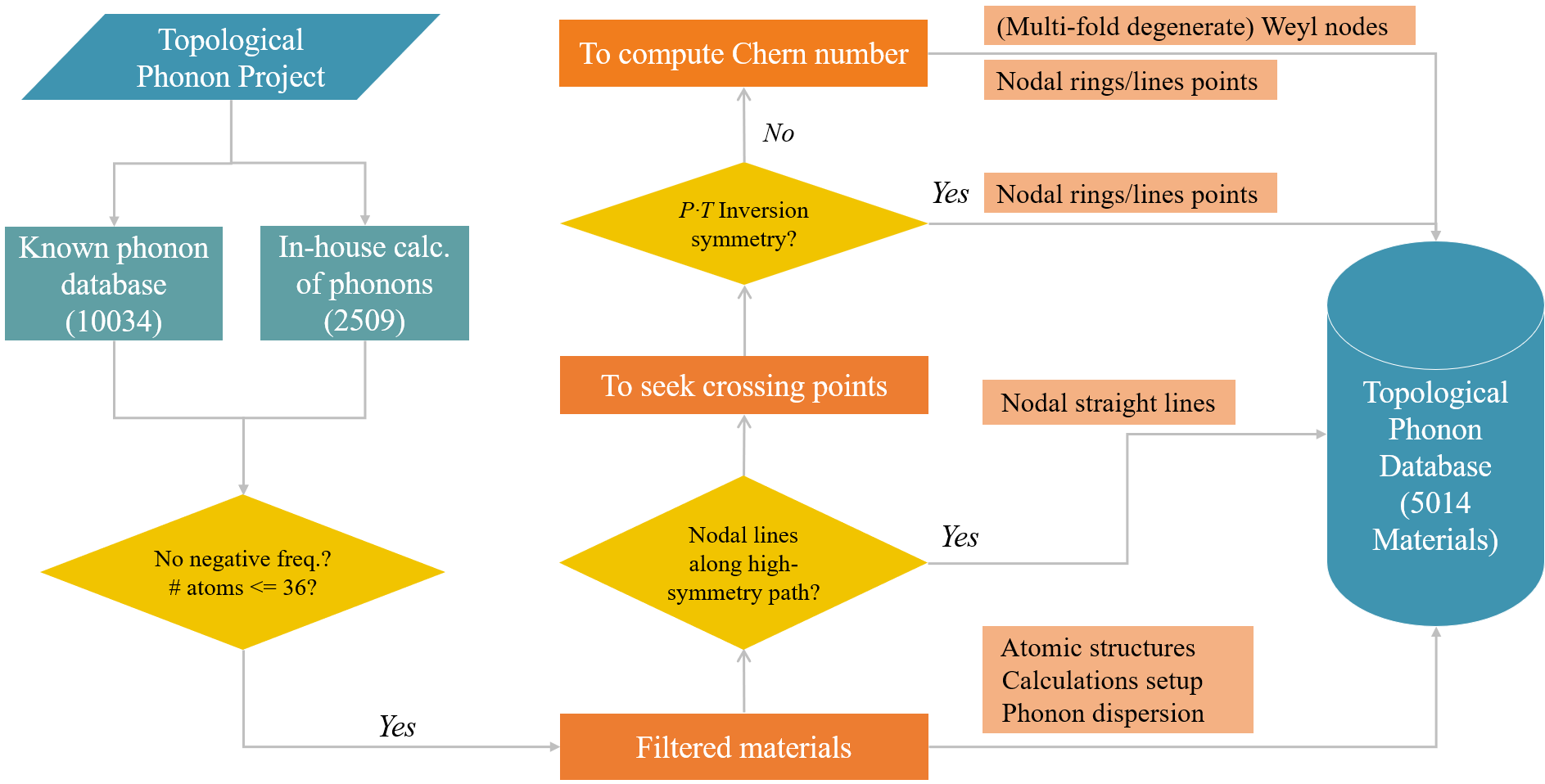
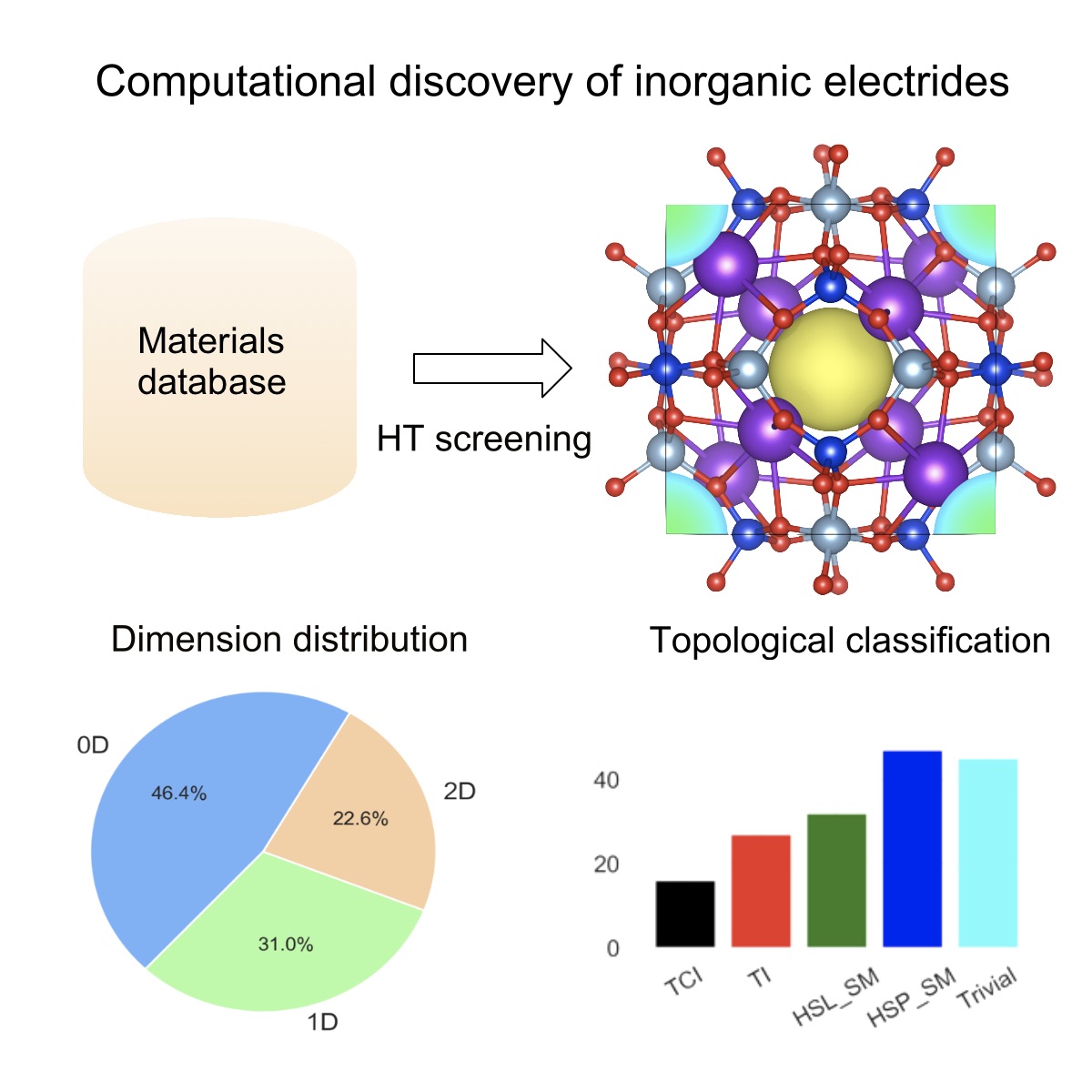
Relevant works
-
Zhu Q, Kang B, Parrish K (2022). Symmetry Relation Database and Its Application to Ferroelectric Materials Discovery. MRS Communications, 12, 686–691 (Early Career Materials Researcher special issue) (arXiv: PDF)
-
Li J-X, Liu J-X, Baronett S.A., Li R-H, Wang L., Zhu Q. and Chen X-Q (2020). Computation and data driven discovery of topological phononic materials. (Open Access: PDF)
-
Qu J-Y, Zagaceta D., Zhang W-W, Zhu Q. (2020). High dielectric ternary oxides from crystal structure prediction and high-throughput screening. Sci. Data, 7, 81. (Open Access: PDF)
-
Zhu Q, Frolov T, K. Choudhary (2019) Computational Discovery of Inorganic Electrides from an Automated Screening. Matter, 1. 1293-1303 (PDF Preview)
-
Zhu S-C, Wang L, Qu, J-Y, Wang, J-J, Frolov T, Chen X-Q and Zhu, Q. (2019) Computational Design of Flexible Electrides with Non-trivial Band Topology Phys. Rev. Materials, 3, 024205 (pdf)
-
Wang J-J, Zhu, Q, Wang Z-H, Hosono H. (2019) Ternary Inorganic Electrides with mixed bonding Phys. Rev. B, 99, 064104 (pdf)
-
Qu J-Y, Zhu S-C, Zhang W-W, and Zhu, Q. (2019) Electrides with Dinitrogen Ligands ACS Appl. Mater. Interfaces, 11, 5256-5263 (pdf)